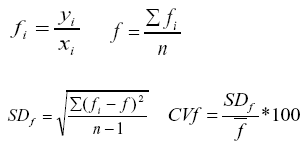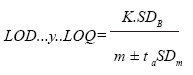
COMUNICACIÓN CORTA
Características metrológicas en la determinación de ácidos urónicos para el control de la calidad en mezclas de oligogalacturónidos
Metrological characteristics in uronic acid determination for quality control of oligogalacturonide mixtures
M.Cs. Yuliem Mederos Torres,I Dra.C. Inés M. Reynaldo Escobar,I Dra.C. Josefa Hormaza Monte-NegroII
IInstituto
Nacional de Ciencias Agrícolas (INCA), gaveta postal 1, San José
de las Lajas, Mayabeque, Cuba, CP 32 700.
IIInstituto Cubano de Investigaciones Azucareras (ICINAZ), carretera
al central Manuel Martínez Prieto km 2½, Ciudad de La Habana,
Cuba.
RESUMEN
La determinación del contenido de ácidos urónicos en el control de la calidad de los oligogalacturónidos es sumamente importante, teniendo en cuenta que ellos están directamente relacionados con la actividad biológica que estos presentan. El objetivo de este trabajo fue realizar el análisis de las características metrológicas de la técnica espectrofotométrica de Blumenkrantz y Asboe-Hansen, en la determinación de ácidos urónicos en mezclas de oligogalacturónidos, empleando como cromóforo el m-hidroxidifenilo. Dentro de las características metrológicas analizadas se encontraron la linealidad, la precisión y los límites de detección y cuantificación. Como resultado el método de Blumenkrantz y Asboe-Hansen se ajustó a un modelo lineal en el rango de concentración de 10-80 µg mL-1. Para el modelo ajustado, el valor del coeficiente de correlación es r= 0,996 y el coeficiente de determinación es R2= 0,992, lo cual indicó muy buena correlación entre la concentración de ácidos urónicos y su absorbancia. La precisión del método se determinó por dos procedimientos, los cuales no sobrepasaron el valor establecido como límite de variabilidad del 5 %. Los límites de detección y cuantificación estuvieron en el orden de 1,6 ± 0,4 µg mL-1 y 5 ± 1 µg mL-1, respectivamente. La concentración de ácidos urónicos se encontró en el orden de los 890 µg mL-1.
Palabras clave: ácido péctico, ácidos urónicos, control de la calidad, oligogalacturónidos.
ABSTRACT
Uronic acid content determination for the quality control of oligogalacturonides is extremely important, considering that they are directly related to their biological activity. The aim of this research was to analyze the metrological characteristics of Blumenkrantz and Asboe-Hansen spectrophotometric method, for uronic acid determination in oligogalacturonide mixtures, by using m-hydroxydiphenyl as chromophore. Linearity, precision and limits of detection and quantification were among the metrological characteristics analyzed. Blumenkrantz and Asboe-Hansen method resulted to be adjusted to a linear model within a concentration range of 10-80 µg mL-1. Regarding such model, the correlation coefficient value is r= 0,996 whereas the determination coefficient is R2= 0,992, indicating a very good correlation between uronic acid concentration and its absorbance. The precision of this method was determined through two procedures, which did not surpass the established value as a variability limit of 5 %. Detection and quantitation limits were in the order of 1,6 ± 0,4 µg mL-1 and 5 ± 1 µg mL-1, respectively. Uronic acid concentration was recorded in the order of 890 µg mL-1.
Key words: pectic acid, uronic acids, quality control, oligogalacturonides.
INTRODUCCIÓN
Los oligogalacturónidos
están compuestos por oligómeros lineales de ácido D-galacturónico
unidos por enlaces del tipo a (1- 4), que pueden ser obtenidos por hidrólisis
ácida o enzimática de los ácidos pécticos (1). Los
oligogalacturónidos han demostrado tener una variada actividad biológica
en las plantas, dentro de las cuales se encuentran la promoción de procesos
morfogenéticos en especies vegetales (2) y la estimulación del
crecimiento vegetal (3), entre otras. Dicha actividad biológica está
determinada por su grado de polimerización, la especie de planta y la
concentración a la cual son aplicados. También ha sido estudiado
su empleo en el secuestro de metales pesados (4). En este sentido, se ha estudiado
el uso de los oligogalacturónidos en la disminución de la toxicidad
del ión Cu2+ en plántulas de tomate, produciendo cambios
en el patrón de absorción de este metal (5).
El Instituto Nacional de Ciencias Agrícolas (INCA), cuenta con una tecnología
patentada a nivel nacional para la obtención de mezclas de oligogalacturónidos.
El control de la calidad de estas mezclas incluye la determinación del
contenido de ácidos urónicos, azúcares neutros y azúcares
reductores presentes en este. Dentro de estas características nombradas,
el contenido de ácidos urónicos es uno de los más importantes,
pues está estrechamente relacionado con la actividad biológica
que estos oligosacáridos presentan en las plantas. Dicha actividad biológica
puede estar basada en la capacidad de los oligogalacturónidos de formar
un enrejado molecular con los iones Ca2+ nombrado caja de huevo (6).
Esta conformación es posible, debido a las múltiples cargas negativas
que presentan los oligogalacturónidos, desde el punto de vista estructural
y que depende de su grado de polimerización.
La evaluación del contenido de ácidos urónicos, en forma
de ácido galacturónico, puede ser realizada mediante la evaluación
espectrofotométrica. Para ello, los métodos espectrofotométricos
más conocidos en esta determinación son aquellos que emplean como
cromóforos al m-hidroxidifenilo (7, 8, 9, 10), el 3,5-dimetilfenol (11,
12) y el carbazol (13, 14). Más recientemente se ha empleado la cromatografía
líquida de alta resolución (15, 16) y la electroforesis capilar
zonal, como método de cuantificación de ácido galacturónico
con detección por UV (17).
De manera general, el procedimiento que emplea el m-hidroxidifenilo como cromóforo
(7) es el más citado de todos y se caracteriza por ser cuantitativo,
sensible y específico, razones por las cuales se seleccionó como
técnica de control de la calidad de las mezclas de oligogalacturónidos.
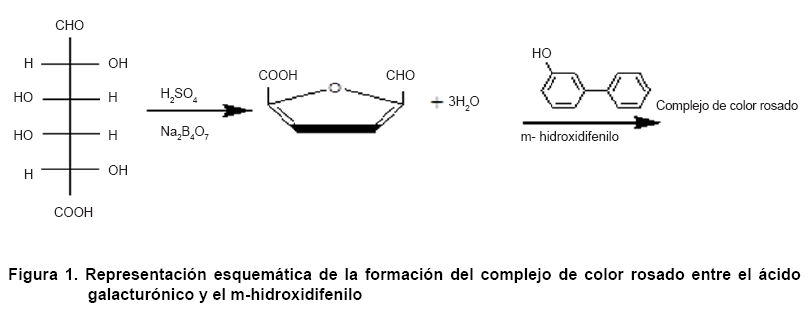
Este método se basa en la aparición de un cromógeno de
color rosado cuando el ácido urónico es calentado a 100 oC
en una mezcla de ácido sulfúrico/tetraborato de sodio y posteriormente
tratado con m-hidroxidifenilo (Figura 1).
El objetivo de este trabajo fue la evaluación de algunas características metrológicas en la técnica de determinación de ácidos urónicos, empleando como cromóforo el m-hidroxidifenilo y de esta forma contribuir a su validación posterior.
MATERIALES Y MÉTODOS
La mezcla de oligogalacturónidos se obtuvo por hidrólisis enzimática (18), con el empleo de ácido poligalacturónico (SIGMA) y el preparado pectinolítico Pectinase obtenido a partir del hongo Aspergillus aculeatus de la corporación Novozyme A/S. La determinación del contenido de ácidos urónicos se realizó de acuerdo al método propuesto por Blumenkrantz y Asboe-Hansen (7), empleando como patrón ácido galacturónico en el rango de concentraciones de 10, 20, 40, 60 y 80 µg mL-1. Las lecturas se realizaron a 520 nm en un espectrofotómetro UV visible Genesys 6 de la corporación Thermo Electron. Las características metrológicas analizadas fueron la linealidad, la precisión y los límites de detección y cuantificación del método (19, 20). La linealidad (n= 3) se determinó por el método de los mínimos cuadrados, con la evaluación de la pendiente, el intercepto, los coeficientes de correlación y determinación, así como la existencia de falta de ajuste, el análisis del gráfico de residuos y el coeficiente de variación del factor respuesta (CVf); este último se determinó a partir de la siguiente secuencia de ecuaciones.
donde:
f es el factor respuesta, yi y xi son la absorbancia
y la concentración del ácido galacturónico, respectivamente,
SDf es la desviación estándar del factor respuesta,
CVf es el coeficiente de variación del factor respuesta y n
el número de réplicas.
La precisión se determinó en términos de repetibilidad,
manteniendo el mismo procedimiento de medición, el mismo operador y sistema
de medición, así como iguales condiciones de operación,
con igualdad de réplicas, en un corto período de tiempo (21).
La precisión se expresó mediante el CV y se determinó como
el cociente entre la desviación estándar y el valor medio del
factor respuesta, expresado en porciento. La precisión se evaluó
por dos vías, mediante la determinación del CV (a partir del análisis
de tres niveles de concentración de ácido galacturónico
10, 40 y 80 µg mL-1) y mediante el análisis de seis
muestras de oligogalacturónidos en condiciones de repetibilidad. Para
el análisis de la mezcla de oligogalacturónidos se realizaron
diluciones de estas a 1000 µg mL-1 en agua desionizada. Los
límites de detección (LOD) y cuantificación (LOQ) se determinaron
mediante las fórmulas de Long and Winefordner (22).
donde:
K= 3 para LOD y 10 para LOQ
SDB= desviación estándar de la absorbancia del blanco
m es la media de la pendiente de las curvas de calibración
ta= distribución t Student para a= 0,01
SDm= desviación estándar de las pendientes de las curvas
de calibración
Los límites se calcularon a partir del análisis de siete blancos.
RESULTADOS Y DISCUSIÓN
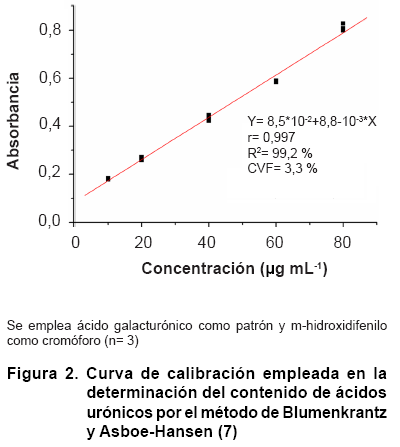
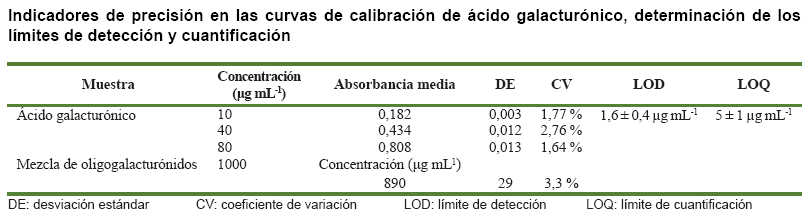
El análisis de la linealidad de la técnica de determinación de los ácidos urónicos se realizó a través de su curva de calibración en el rango de concentración de 10-80 µg mL-1 (Figura 2). El análisis de la regresión mostró que se ajustó a un modelo lineal descrito por la ecuación de primer grado y= 0,09 + 8,8.10-3x para el rango de concentraciones especificado. Para el modelo ajustado, el valor del coeficiente de correlación es r= 0,996172, mientras que el coeficiente de determinación es R2= 0,992, lo cual indica una excelente correlación entre la concentración y la absorbancia. El análisis de significación de la pendiente mostró que la probabilidad de error es menor que 0,05 (P= 0,0000), lo cual indicó que esta es significativamente diferente de cero. Por otra parte, el análisis de significación del intercepto mostró que la probabilidad de cometer error, determinado a partir de la prueba t de Student es menor que 0,05 (P= 0,0000), lo cual indica que el intercepto (0,09) debe ser tenido en cuenta al realizar los análisis. El coeficiente de variación del factor respuesta (CVf) en el rango de concentraciones analizado fue de 3,3 %, el cual es menor que 5 %, valor conocido como límite de variabilidad, por lo cual se considera adecuado (23).
La prueba de falta de ajuste verifica si el modelo seleccionado (modelo lineal) es adecuado para describir los datos observados o si debe ser utilizado un modelo más complejo. La probabilidad (P= 0,0012) asociada al valor de F de la falta de ajuste resultó menor que 0,05, por lo que hay una falta de ajuste al modelo lineal estadísticamente significativo para un nivel de confianza del 95 % (Tabla).
Aun cuando es
conocido que el análisis de la linealidad en un método de determinación
espectrofotométrico se realiza a partir de la búsqueda de la ecuación
lineal que lo describa en un rango de concentraciones especificado, el empleo
del programa estadístico STATGRAPHICS PLUS 5.1 (24) permite
verificar, a partir de modelos curvilíneos alternativos, qué modelo
matemático describe mejor el sistema.
El modelo ajustado que mejor describió al sistema fue raíz cuadrada
con un R2= 99,41 %; sin embargo, el modelo lineal solo difirió
de este en un 0,17 %, lo que concuerda con la falta de ajuste encontrada en
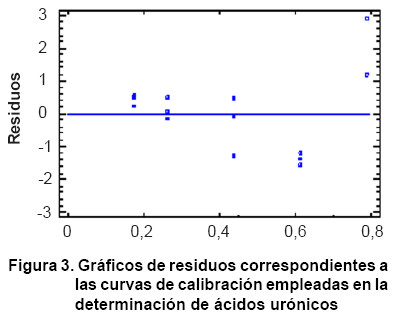
el análisis de varianza. Por otra parte, el gráfico de residuos
mostró que los resultados presentan una distribución aleatoria,
lo cual confirma que el modelo escogido es el adecuado (Figura
3).
La evaluación
de la linealidad en un método espectrofotométrico se basa en la
propiedad de las sustancias de absorber la luz y debe cumplir con la Ley de
Lambert-Beer, que plantea que la luz absorbida es directamente proporcional
a la concentración de la sustancia (25). Por tanto, se puede decir que
esta relación quedó descrita a partir de la ecuación lineal
de primer grado, y= 0,09 + 8,8.10-3x, cuyos coeficientes de correlación
y determinación demostraron la proporcionalidad existente entre la concentración
y absorbancia, cumpliendo así con la Ley de Lambert-Beer.
La precisión del método se evaluó en términos de
repetibilidad. La determinación del coeficiente de variación a
tres niveles de concentración diferentes mostró que este oscila
entre el 1,64 y 2,76 %, el cual se encuentra por debajo del valor establecido
como límite de variabilidad (5 %), por lo que el método se considera
adecuado (Tabla).
La determinación del límite de detección y cuantificación
en la curva de calibración (Tabla) mostró que
el nivel más bajo de ácidos urónicos que puede ser detectado
mediante esta técnica es de 1,6 ± 0,4 µg mL-1.
A su vez, la concentración más baja que puede ser determinada
con precisión es 5 ± 1 µg mL-1.
Los valores de limites de detección y cuantificación calculados
indican que el método de determinación de ácidos urónicos
es adecuado, teniendo en cuenta que la concentración de ácidos
urónicos presente en muestras de mezclas de oligogalacturónidos
está en el orden de los 890 µg mL-1, siendo estos los
componentes mayoritarios, representando aproximadamente el 89 % de las muestras.
Dicha concentración está en el orden de los valores indicados
en la literatura en la caracterización de ácido péctico
comercial (SIGMA) de origen cítrico (26), donde la concentración
de ácidos urónicos representó el 87 % de la muestra analizada.
La determinación del límite de detección permitió
referir que la respuesta lineal del método comprendió el rango
de 1,6 a 80 µg mL-1. Este resultado se encontró en el
orden de lo informado por otros autores (27), al realizar la determinación
espectrofotométrica de los ácidos urónicos en muestras
de té verde, donde la respuesta lineal del método oscila entre
1 y 100 µg mL-1.
CONCLUSIONES
De manera general y a modo de conclusión, teniendo en cuenta todas las características metrológicas analizadas anteriormente, se puede decir que la técnica de determinación de ácidos urónicos, de acuerdo al método propuesto por Blumenkrantz y Asboe-Hansen (7), puede ser empleado como técnica de control de la calidad en mezclas de oligogalacturónidos, puesto que cumple con el criterio de linealidad, precisión y límites de detección y cuantificación adecuados para este fin.
BIBLIOGRAFÍA
1. Mederos, T.
Y.; Hormaza, M. J.; Reynaldo, E. I. y Montesino, S. R. ‘‘Caracterización
de mezclas de oligogalacturónidos bioactivos’’. Revista CENIC. Ciencias
Químicas, vol. 42, no. 2-3, 2011, pp. 1–5, ISSN 1015-8553.
2. Terry, A. E.; Ruiz, P. J.; Tejeda, P. T. y Reynaldo, E. I. ‘‘Efectividad
agrobiológica del producto bioactivo Pectimorf® en el
cultivo del Rábano (Raphanus sativus L.)’’. Cultivos Tropicales,
vol. 35, no. 2, junio de 2014, pp. 105-111, ISSN 0258-5936.
3. Alvarez, B. I.; Reynaldo, E. I.; Cartaya, R. O. y Teheran, Z. ‘‘Efectos de
una mezcla de oligogalacturónidos en la morfología de hortalizas
de importancia económica’’. Cultivos Tropicales, vol. 32, no.
3, septiembre de 2011, pp. 52-57, ISSN 0258-5936.
4. Ozturk, S.; Aslim, B.; Suludere, Z. y Tan, S. ‘‘Metal removal of cyanobacterial
exopolysaccharides by uronic acid content and monosaccharide composition’’.
Carbohydrate Polymers, vol. 101, 30 de enero de 2014, pp. 265-271,
ISSN 0144-8617, DOI 10.1016/j.carbpol.2013.09.040.
5. Cartaya, O. E.; Reynaldo, I.; Peniche, C. y Garrido, M. L. ‘‘Empleo de polímeros
naturales como alternativa para la remediación de suelos contaminados
por metales pesados’’. Revista Internacional de Contaminación Ambiental,
vol. 27, no. 1, febrero de 2011, pp. 41-46, ISSN 0188-4999.
6. Cabrera, J. C.; Cambier, P. y Cutsem, P. ‘‘Drug encapsulation in pectin hydrogel
beads. A systematic study of simulated digestion media’’. International
Journal of Pharmacy and Pharmaceutical Sciences, vol. 3, 2011, pp. 292–299,
ISSN 0975-1491.
7. Blumenkrantz, N. y Asboe-Hansen, G. ‘‘New method for quantitative determination
of uronic acids’’. Analytical Biochemistry, vol. 54, no. 2, agosto
de 1973, pp. 484-489, ISSN 0003-2697, DOI 10.1016/0003-2697(73)90377-1.
8. Caesar-TonThat, T.; Sainju, U. M.; Wright, S. F.; Shelver, W. L.; Kolberg,
R. L. y West, M. ‘‘Long-term tillage and cropping effects on microbiological
properties associated with aggregation in a semi-arid soil’’. Biology and
Fertility of Soils, vol. 47, no. 2, 18 de noviembre de 2010, pp. 157-165,
ISSN 0178-2762, 1432-0789, DOI 10.1007/s00374-010-0508-2.
9. Li, T.; Zhu, R.; Dong, Y.; Liu, Y.; Li, S. y Chen, G. ‘‘Effects of Pectin
Pentaoligosaccharide from Hawthorn (Crataegus pinnatifida Bunge. var.
Major) on the Activity and mRNA Levels of Enzymes Involved in Fatty Acid Oxidation
in the Liver of Mice Fed a High-Fat Diet’’. Journal of Agricultural and
Food Chemistry, vol. 61, no. 31, 7 de agosto de 2013, pp. 7599-7605, ISSN
0021-8561, DOI 10.1021/jf400283w.
10. Parwani, L.; Bhatnagar, M.; Bhatnagar, A. y Sharma, V. ‘‘Antioxidant and
iron-chelating activities of cyanobacterial exopolymers with potential for wound
healing’’. Journal of Applied Phycology, vol. 26, no. 3, 6 de noviembre
de 2013, pp. 1473-1482, ISSN 0921-8971, 1573-5176, DOI 10.1007/s10811-013-0180-7.
11. Luzio, G. A. ‘‘Determination of galacturonic acid content of pectin using
a microtiter plate assay’’. Proceedings of the Florida State Horticultural
Society, vol. 117, 2004, pp. 416–421, ISSN 0097-1219.
12. Silva, T. C. F.; Colodette, J. L.; Lucia, L. A.; Oliveira, R. C. de; Oliveira,
F. N. y Silva, L. H. M. ‘‘Adsorption of Chemically Modified Xylans on Eucalyptus
Pulp and Its Effect on the Pulp Physical Properties’’. Industrial &
Engineering Chemistry Research, vol. 50, no. 2, 19 de enero de 2011, pp.
1138-1145, ISSN 0888-5885, DOI 10.1021/ie101960a.
13. Jing, Y.; Zhu, J.; Liu, T.; Bi, S.; Hu, X.; Chen, Z.; Song, L.; Lv, W. y
Yu, R. ‘‘Structural Characterization and Biological Activities of a Novel Polysaccharide
from Cultured Cordyceps militaris and Its Sulfated Derivative’’. Journal
of Agricultural and Food Chemistry, vol. 63, no. 13, 8 de abril de 2015,
pp. 3464-3471, ISSN 0021-8561, DOI 10.1021/jf505915t.
14. Li, W.; Xia, X.; Tang, W.; Ji, J.; Rui, X.; Chen, X.; Jiang, M.; Zhou, J.;
Zhang, Q. y Dong, M. ‘‘Structural Characterization and Anticancer Activity of
Cell-Bound Exopolysaccharide from Lactobacillus helveticus MB2-1’’. Journal
of Agricultural and Food Chemistry, vol. 63, no. 13, 8 de abril de 2015,
pp. 3454-3463, ISSN 0021-8561, DOI 10.1021/acs.jafc.5b01086.
15. Nagel, A.; Sirisakulwat, S.; Carle, R. y Neidhart, S. ‘‘An Acetate–Hydroxide
Gradient for the Quantitation of the Neutral Sugar and Uronic Acid Profile of
Pectins by HPAEC-PAD without Postcolumn pH Adjustment’’. Journal of Agricultural
and Food Chemistry, vol. 62, no. 9, 5 de marzo de 2014, pp. 2037-2048,
ISSN 0021-8561, DOI 10.1021/jf404626d.
16. Zhang, Y.; Zhang, P.; Wang, Z. y Huang, L. ‘‘An Innovative Derivatization
Method for Simultaneous Determination of Uronic Acids and Neutral and Amino
Sugars in Coexisting Samples by Hplc-Esi-Ms/Ms2’’. Journal of
Liquid Chromatography & Related Technologies, vol. 34, no. 16, 1 de
octubre de 2011, pp. 1754-1771, ISSN 1082-6076, DOI 10.1080/10826076.2011.579216.
17. Xia, Y.; Liang, J.; Yang, B.; Wang, Q. y Kuang, H. ‘‘A new method for quantitative
determination of two uronic acids by CZE with direct UV detection’’. Biomedical
Chromatography, vol. 25, no. 9, 1 de septiembre de 2011, pp. 1030-1037,
ISSN 1099-0801, DOI 10.1002/bmc.1564.
18. Cabrera, J.; Iglesias, R. y Hormaza, J. Procedimiento de obtención
de una mezcla de oligosacáridos pécticos estimuladora del enraizamiento
vegetal. no. 22859, Inst. Oficina Cubana de la Propiedad Industrial, La
Habana, Cuba, 2003.
19. Ortega, G. M.; Rodríguez, M. C. y Zhurbenko, R. ‘‘Validación
de métodos alternativos para análisis microbiológico de
alimentos y aguas. Métodos cualitativos’’. Revista Cubana de Higiene
y Epidemiología, vol. 48, no. 2, agosto de 2010, pp. 162-176, ISSN
1561-3003.
20. Perdomo, R. R.; Otero, M. G. y González, M. ‘‘Validación del
método del sulfato de brucina para la determinación de nitrógeno
forma de nitrato en aguas superficiales’’. Revista Cubana de Química,
vol. 24, no. 2, 2012, pp. 155–165, ISSN 2224-5421.
21. ISOI. International Vocabulary of Metrology. Basic and general concepts
and associated terms. No. ISO/IEC 99: 2007, Geneva, Switzerland, 2007,
p. 92.
22. Long, G. L. y Winefordner, J. D. ‘‘Limit of detection. A closer look at
the IUPAC definition’’. Analytical Chemistry, vol. 55, no. 7, 1 de
junio de 1983, p. 712A-724A, ISSN 0003-2700, DOI 10.1021/ac00258a001.
23. Oficina Nacional de Normalización. Guía para la validación
de métodos de ensayo químicos para alimentos. No. NC TS 368:
2010, La Habana, Cuba, 2010, p. 48.
24. Statistical Graphics Crop. STATGRAPHICS® Plus [en
línea]. (ser. Profesional), versión 5.1, [Windows], 2000, Disponible
en: <http://www.statgraphics.com/statgraphics/statgraphics.nsf/pd/pdpricing>.
25. Owen, T. Fundamentals of Modern UV-Visible Spectroscopy: A Primer.
1a ed., edit. Agilent Technologie, German, 2000, 136 p., Publication
number 5980-1397.
26. Cabrera, J. C.; Gutiérrez, A. y Varela, M. ‘‘Comparación del
ácido péctico obtenido a partir de albedo de lima persa con el
polipectato de sodio reactivo’’. Cultivos Tropicales, vol. 16, no.
3, 1995, pp. 29–31, ISSN 1819-4087.
27. Chen, H.; Zhang, M. y Xie, B. ‘‘Quantification of Uronic Acids in Tea Polysaccharide
Conjugates and Their Antioxidant Properties’’. Journal of Agricultural and
Food Chemistry, vol. 52, no. 11, 1 de junio de 2004, pp. 3333-3336, ISSN
0021-8561, DOI 10.1021/jf0349679.
Recibido: 17 de
diciembre de 2014
Aceptado: 28 de julio de 2015
M.Sc. Yuliem Mederos Torres, Instituto Nacional de Ciencias Agrícolas (INCA), gaveta postal 1, San José de las Lajas, Mayabeque, Cuba, CP 32 700. Email: yuliem@inca.edu.cu